Tuesday, December 29, 2009
Salting out and in
My latest story for Physical Review Focus concerns calculations of the tendency of various ions dissolved in water to accumulate at its surface.
This is a really old problem, discussed by some of the giants of physical chemistry. In the 1930s, for example, later Nobel winner Lars Onsager and others suggested that the termination of the electrical polarization at the surface of the water would give rise to an "image charge"--a surface charge of the same sign as the ion that creates an electric field just like that of a point charge at the mirror-image location on the other side of the interface. The repulsion from this image charge, they suggested, would keep ions away from the surface.
People have apparently suspected for decades that things can't be that simple, because different ions alter the surface tension to different degrees, indicating that they are changing the energy of the surface, presumably by being part of it. But only in the past decade or so have new experiments and simulations shown that some simple negative ions like halogens can be stable at the surface. Such ions at the surface of atmospheric droplets could be important catalysts, for example for breaking down ozone.
The two closely related Physical Review Letters that motivated the Focus story attribute the attractiveness of the surface position of a large negative ion to its internal polarizability. The internal rearrangement of charge, they say, allows the ion to retain much of the electrostatic attraction to nearby water molecules without creating a big hole in the water. However, I talked to another researcher who attributes the stabilization of the surface ion to a distortion it induces in the shape of the nearby surface. These both seem like potentially important effects, and both may play a role in the ultimate understanding.
The difference between the two could be important, though, for a related and even older phenomenon: the effect of various added salts on dissolved proteins. In 1888, Hofmeister ranked a series of ions in terms of their effectiveness in precipitating the proteins, and the order of the series mirrors that which was later found for the effects of ions on surface tension.
"Salting out" occurs when an added salt reduces the solubility of a protein, presumably by tying up water molecules and raising its effective concentration. This effect has been used for decades to create the protein crystals needed for structural studies like x-ray crystallography.
In contrast, "salting in" makes the protein more soluble, but may denature it. Salts that have this effect may alter the repulsion between water and the hydrophobic regions of the protein. This repulsion is critical for maintaining the shape of proteins that naturally occur in the bulk of the cell, since that shape generally presents hydrophilic regions to the solution and shelters hydrophobic regions inside. (Proteins that naturally occur in membranes, by contrast, generally expose a hydrophobic stripe where they are embedded in the non-aqueous center of the membrane sheet.)
The polarizability of ions at the protein-water interface could have an important effect on this repulsion. In contrast, since the water-protein interface is entirely within the liquid, changing the shape of the interface wouldn't seem to be an option.
It is true that many proteins take on their final shapes only in the presences of "chaperone" proteins, which can also help fix them up if they become denatured. Nonetheless, any insight into the interactions between water and proteins could be very important to understanding why they fold the way they do, and how circumstances might change that folding.
Monday, December 14, 2009
Rules to Design By
Once something gets really too complicated, it's almost certain to fail. So how can computer chips, with their billions of components, work at all?
We know lots of other complicated systems, like the world economy or our own bodies. And we know those systems fail often dramatically or tragically.
Of course, computers fail, too, as you know if you've seen a "blue screen of death" recently. But although it won't make you feel any better, those crashes almost always arise from problems with the software, not the hardware it runs on.
So how do engineers ensure that integrated circuits, diced by the score from semiconductor wafers, have a very good chance of working?
Design rules.
Simply put, design rules are a contract between the engineers designing the process for making chips and the engineers designing circuits to put on them. The process engineers guarantee that, if the circuit designers follow these rules in the geometry of their circuits, the chips will work (most of the time).
You may have heard "minimum design rule" used as a shorthand to describe a particular "generation" of computer chips, such as the "32nm" technology recently introduced by Intel. But that is shorthand is somewhat misleading.
For one thing, the true gate length of the transistor--which is critical to their speed and power--is generally about half the generation name. In addition, the "coded" gate length--the length in computer-aided-design files--is not usually the smallest design rule. And this is just one of hundreds of rules that are required to define a technology.
Rather than dive into the details of transistor geometry, consider a simpler design rule: the minimum width of a metal wire connecting the transistors. Together with the spacing between the wires, this dimension determines how tightly the wiring can be packed, which for some circuits determines how many transistors can be used in a parcel of semiconductor real estate.
The minimum safe design width of a wire depends on how fine it can be made and still assure that it will conduct electricity. This has to be guaranteed even under variations over time of the process used to make it, as well as the variation in that process across a, say, 12-inch diameter wafer.
To test what number is safe, the process engineers will make a whole series of test patterns, each consisting of very long wires with various design widths. After measuring hundreds of these test structures, they have a good idea what they can reliably make.
In developing the process technology, they have hundreds of test structures, each aimed at testing one or more design rules. The structures are automatically measured on different positions on different wafers made in different processing runs. Only then will the engineers have the confidence to guarantee that any circuit that follows those rules will work.
After a long process, a set of design rules will be given to designers to use for their circuit layouts. None of this would work without computers to check whether a particular chip layout meets the rules, since the job is beyond human capacity. Therefore a key feature of the design rules is that they can be embodied in an efficient algorithm.
The design-rule paradigm has been extraordinarily successful. But its success depends on a characteristic of the failures it is intended to prevent: they are all dependent on the local properties of the circuit. Some of the more complex rules involve quantities like the area of the metal "antenna" that is connected to a particular device at some point during processing. And frequently the engineers will play it safe by crafting the rules to cover the worst possible situation. But if the rules are chosen and followed properly, there is no chance for a combination of small choices that satisfy the rules to join together to cause a problem in the larger circuit. That's what makes a chip with a billion transistors possible.
Friday, December 11, 2009
Chromatin Compartments
The fractal packing of some DNA is just one of the interesting results from the recent Science paper by Lieberman-Aiden and colleagues. Of greater practical importance is the ability of their experimental technique to assign each region of DNA to one of two compartments.
The fact that some DNA regions, called heterochromatin, are packed more densely than other regions, called euchromatin, was discovered 80 years ago, by observing darker and lighter regions of stained nuclei under the optical microscope. Researchers have since learned that the heterochromatin is more densely packed, and that the genes it contains are transcriptionally silent. Heterochromatin also tends to segregate to the periphery of the nucleus, but to avoid the nuclear pores through which gene products are exported.
The Science authors did not mention this well-known classification. However, when they measured which regions of the genome were close together in the clumped DNA, they found that they could divide the mappable regions of the genome into two distinct "compartments." Regions from compartment A were more likely to lie close to other regions from compartment A, and similarly for compartment B. Importantly, they could make this assignment even for regions on different chromosomes, suggesting that the compartments represent regions of the nucleus in which segments of different chromosomes mingle.
The researchers also found that regions in compartment B were much more likely to be in close contact, so they designated that compartment "closed," and the other one "open." But Erez Lieberman-Aiden told me that "it seemed best to use terminology attached to things that we can probe and which clearly correspond to our compartments." Indeed, the regions they call "open" correspond well to the regions that can are accessible to DNA-digesting enzymes, but do not correspond to the light and dark bands that appear on the chromosomes during cell division.
Although the relationship to microscopically-observed partitioning may need clarification, the ability to globally map closed and open regions of the genome could be a very powerful tool. Looking at different cell types, for example, could reveal overall "signatures" in the chromosome arrangements. Such cell-type-specific patterns are already known to exist in the arrangement of histone modifications, which affect the nucleosome arrangement.
In addition, the chromatin structure enters into regulation of individual genes. Enhancer elements in the DNA sequence, for example, can affect the expression of quite distant genes, while an intervening insulator region can block that effect. Models of these influences generally involve large loops of DNA, but some also include the notion of a densely-packed and transcriptionally silent "scaffold" region that is reminiscent of the closed compartment. Determining which sections of the sequence are in the closed or open arrangements, especially in cells with different types of activity, could add some much-needed experimental visibility into the regulatory activity of these critically important elements.
[For physicist readers: as I was wrapping up this entry, the latest Physics Today arrived with a news story on this subject.]
Thursday, December 10, 2009
Fractal DNA
Packing meters of DNA into a nucleus with a diameter a million times smaller is quite a challenge. Wrapping the DNA around nucleosomes, and arranging these nucleosomes into 30nm fibers, both help, but these structures must themselves be packed densely. Beautiful new research, reported in Science in October, supports a 20-year old idea that some DNA is arranged in an exotic knot-free fractal structure that is particularly easy to unpack.
Alexander Grosberg, now at New York University, predicted (1M pdf) in 1988 that a polymer would initially collapse into a "crumpled globule," in which nearby segments of the chain would be closer to each other than they would be in the final, equilibrium globule. Creating the equilibrium structure requires "reptation," in which the polymer chain threads its way through its own loops, forming knots. This gets very slow for a long chain like DNA. Grosberg also applied (1M pdf) these ideas to DNA, and explored whether fractal patterns in the sequence could stabilize it. But experimental evidence was limited.
Now Erez Lieberman-Aiden and his coworkers at MIT and Harvard have devised a clever way to probe the large-scale folding structure of DNA, and found strong support for this picture.
The experiment is similar to chromatin immunoprecipitation techniques that look for DNA regions that are paired to target proteins by crosslinking and precipitating the pairs and then sequencing the DNA. In this case, however, the researchers crosslink nearby sections of the collapsed DNA to each other. To sequence both sections of DNA, they first splice the ends of the pairs to each other to form a loop, and then break them apart at a different position in the loop. The result is a set of sequence pairs that were physically adjacent in the cell; their positions along the DNA are found by matching them to the known genome.
The researchers found that the number of neighboring sequences decreases as a power law of their sequence separation, with an exponent very close to -1, for sequence distances in the range of 0.5 - 7 million bases. This is precisely the expected exponent for the crumpled--or fractal--globule. This structure is reminiscent of the space-filling Peano curve with its folds, folds of folds, and folds of folds of folds forming a hierarchy. In contrast, the equilibrium globule has an exponent of -3/2.
As a rule, I don't put a lot of stock in claims that a structure is fractal simply by seeing a power law, or a straight line on a double-logarithmic plot, unless the data cover at least a couple of orders of magnitude. After all, a true fractal is self-similar, meaning that the picture looks exactly the same at low resolution at high resolution, and in many cases there's no reason to think that fine structure resembles the coarse structure at all.
But when there's a good theoretical argument for similar behavior at different scales, I relax my standards of evidence a bit. For example, there's a good argument that rate the random walk of a diffusing molecule looks into neighboring volumes looks similar, whatever the size of the volume you consider--this is a known fractal. The standard polymer model is just a self-avoiding random walk, which adds the constraint that two parts of the chain can't occupy the same space. The DNA data are different in detail, but the mathematical motivation is similar.
At the conference I covered last week in Cambridge, MA, Lieberman-Aiden noted that the fractal structure has precisely the features you would want for a DNA library: it is compact, organized, and accessible. The densely packed structure keeps nearby sequence regions close in space, and parts of it can easily be unfolded to allow the transcription machinery to get access to it. Co-author Maxim Imakaev has verified all of these features with simulations of the collapsing DNA.
These experiments and simulations are fantastic, and the fractal globule structure makes a lot of sense. But this dense structure makes it all the more amazing what must happen when cells divide, making a complete copy of each segment of DNA (except the telomeres), and ensuring that the epigenetic markers on the DNA and histones of one copy are replicated on the other. It's still an awesome process.
Monday, December 7, 2009
Short RNAs to the Rescue
Ever since scientists realized, just over a decade ago, that exposing cells to short snippets of RNA could affect the activity of matching genes, they have dreamed if harnessing this RNA interference, or RNAi, to fight diseases. In the past week, two groups have announced progress toward that goal, treating chimpanzees with hepatitis C and mice with lung cancer.
RNAi, which rapidly earned a 2006 Nobel Prize, is just one facet of the many ways in which short RNAs regulate gene activity. Researchers have since found numerous types of naturally occurring short RNA that play important roles in development, stem cells, cancer, and other biological processes. These RNA-based mechanisms could seriously revise the emerging understanding of how cellular processes are controlled.
Over the same period, manipulating genetic activity with short RNAs has become an essential tool in biology labs. Cells process various forms of short RNA, such as short-hairpin RNA (shRNA) and small interfering RNA (siRNA) into RNA-protein complexes that reduce (usually) how much protein is made from a messenger RNA that include a complementary (or nearly complementary) sequence.
This technique gives researchers a quick way to learn about what a particular gene does, at least in culture dishes, sidestepping the laborious creation and breeding of genetically-modified critters. (Or if they do put in the time, they can insert genes that allow them to controllably trigger RNAi to knock down a gene only in particular cells or after it has completed an indispensible task in helping an organism to grow.)
But affecting genetic regulation in patients faces the challenges of "delivery" that are well-known in the pharmaceutical industry: To have a beneficial effect, the short RNA must survive in the body, get inside the right cells in large quantities, and not cause too many other effects in other cells. The New York Academy of Sciences has a regular series on the challenges of using RNA for treatment, and I covered one very interesting meeting in 2008.
Molecular survival is the first challenge. Researchers have developed various chemical modifications that help RNA (or a lookalikes) withstand assaults by enzymes that degrade rogue nucleic acids. Santaris, for example, which helped in the hepatitis project, has developed proprietary modifications it calls "locked nucleic acids," or LNA. Other researchers and companies are exploring similar techniques.
Getting the protected RNA to the right tissue is another challenge. Foreign chemicals are naturally cycled to the liver for processing, so it's fairly easy to target this organ. For this reason, the hepatitis results don't really prove that the technique is useful for other tissues. The Santaris release also neglects to mention any publication associated with the research.
The mouse lung cancer result appears in Oncogene. The lead Yale researcher, Frank Slack, regularly studies short RNAs in the worm C. elegans, as I described in a recent report from the New York Academy of Sciences. In this work, he teamed with Mirna Therapeutics, which aims to use the short-RNA-delivery vehicle to replace naturally occurring microRNA that are depleted in cancer, like the let-7 they used for this study. The mouse cancers did not disappear, but they regressed to about a third of their previous size, according to the release. Mirna says that since they are replacing natural microRNAs, their technique shouldn't induce many side effects in other tissues.
A further risk for small-RNA delivery is immune responses. The field of gene therapy is only now recovering from the 1998 death of Jesse Gelsinger in what looks like a massive immune response to the virus used to insert new genes in his cells. Although the short-RNA response will be different, some cellular systems are primed to respond to the foreign nucleic acids brought in by viruses.
It's likely that there will be many twists and turns along the way, and I haven't solicited expert opinions on these studies, but they seem to be intriguing steps toward the goal of using RNA not just to study biology, but to change people's lives.
Wednesday, December 2, 2009
Massachusetts Dreaming
Today I'm taking Amtrak to Cambridge--our fair city--MA, for an exciting back-to-back-to-back trio of conferences at the MIT/Harvard Broad (rhymes with "road") Center.
Two of the conferences are described as satellites to RECOMB (Research in Computational Molecular Biology), even though that meeting was in Tucson in May. One of these is on regulatory genomics and the other on systems biology. The third is the fourth meeting of the DREAM assessment of methods for modeling biological networks, a series I've covered since its organizational meeting at the New York Academy of Sciences in 2006.
There's a lot in common between these conferences, so it's not always easy to notice the boundaries. The most tightly focused is DREAM--Dialog on Reverse-Engineering Assessment and Methods. The goal is simple to state: what are the best ways to construct networks that mimic real biological networks, and how much confidence should we have in the results. In practice, things are not so straightforward, and border on the philosophical question of how to distinguish models and "reality." The core activity of DREAM is a competition to build networks based on diverse challenges.
The Regulatory Genomics meeting covers detailed mechanisms of gene regulation, often focusing on more formal and algorithmic aspects than would be expected in a pure biology meeting. The Systems Biology meeting addresses techniques, usually based on high-throughput experimental tools, for attacking large networks head on, rather than taking the more traditional pathway-by-pathway approach.
I'll be writing synopses of the invited talks and the DREAM challenges for an eBriefing at NYAS, but I'll be free to relax and enjoy the contributed talks and posters. This promises to be a rich and exhausting five days.
Tuesday, December 1, 2009
Packing DNA Beads
The dense packing of DNA in the nucleus of eukaryotes strongly affects how genes within it are expressed, with some regions much more accessible to the transcription machinery than others. At the shortest scales, the accessibility of the DNA double helix is reduced where it is wound around groups of eight histone proteins to form nucleosomes, and the precise position of the nucleosomes in the sequence affects which genes are active.
At a slightly larger scale, the nucleosomes are rather closely packed along the DNA. They can remain floppy, like beads on a string, or they can fold into rods of densely packed beads, which further reduces the accessibility of their DNA. Other proteins in the nucleus, notably the histone H1, help to bind together this dense packing. These rods can pack further, with the help of other proteins.
The histone proteins that form the core of the nucleosome, two copies each of H2A, H2B, H3, and H4, have stray "tails" extending from the core. Small chemical changes at particular positions along these tails can have surprisingly large influence on the expression of the associated DNA. For example, the modification H3K27me3 (three methyl groups attached to the lysine at position 27 on the tail of histone H3) represses expression, while acetylation of the same amino acid, H3K27ac activates expression. There is also a more substantial modification, in which histone H2A is replaced by a variant called H2A.Z also modifies expression.
The detailed mechanisms by which the modifications affect expression, such as changing the wrapping of nucleosomes, the packing of nucleosomes, or recruiting of other proteins in the nucleus, are areas of active research.
Since there are dozens of possible histone tail modifications, there are vast numbers of possible combinations of modifications. Some researchers have proposed that these combinations could each prescribe different expression patterns, for example during development. However, the evidence for a combinatorial "histone code" analogous to the three-base codons of the genetic code remains weak.
Nonetheless, proteins that can modify the tails, either adding or removing a chemical group, can have lasting effects on the activity of the underlying genes. The sirtuin proteins that are candidates for longevity-extending drugs, for example, are best known for their role as histone deacetylases.
Some histone modifications can be passed down through cell division or reproduction, so they qualify as epigenetic changes. In contrast to the natural replication of the mirror-image DNA sequence, replicating histone modifications requires a much more complicated process.
Changes in the pattern of histone modifications are found in many basic biological processes, including development, stem-cell maintenance, and cancer. Particular modification patterns have been used to find specific functional sequences within the DNA, such as transcription start sites and enhancers. For these reasons, the ENCODE project mapped modifications as part of their survey of a select part of the human genome for intense study.
Understanding the mechanisms and roles of DNA organization and how it is changed will be essential to a complete picture of gene regulation.
Monday, November 30, 2009
The Honest Broker
In case you hadn't noticed, discussion of global warming has become somewhat polarized. Amid accusations, on the one hand, that industry-financed non-experts deliberately sow confusion, and on the other that a leftist cabal exaggerates the risks and threatens our economy, Roger A. Pielke, Jr. is something of an anomaly.
A professor of environmental studies at the University of Colorado, Pielke is an expert who endorses the broad consensus that humans are causing dangerous changes. But he also criticizes scientists like those on the Intergovernmental Panel on Climate Change for stifling legitimate dissent in the service of narrow policy options. In his 2007 book, The Honest Broker: Making sense of science in policy and politics, Pielke touches on climate change only tangentially as he outlines how scientists can more constructively contribute to contentious policy decisions.
Reading the title, I thought at first that I understood what Pielke meant by an "Honest Broker." As an undergraduate thirty years ago I dabbled in the still-young academic field of Science, Technology, and Society. Books like Advice and Dissent: Scientists in the political arena, by Joel Primack and Frank Von Hippel illustrated how scientists who step outside their specialized knowledge to advocate particular policies risk both their own credibility and that of science. To preserve the authority of expertise, scientists should be careful and clear when they spoke outside of their specialty.
But the intervening years, Pielke says, have shown that the whole notion that science provides objective information that is then handed over to inform policy makers, the so-called linear model, is naïve and unrealistic. Only rarely, when people share goals and the relation between causes and effects is simple, can scientists meaningfully contribute by sticking to their fields of expertise as a "Pure Scientist" or by providing focused answers to policy questions as a "Science Arbiter."
More frequently, people do not share goals and the causal relationships are more complicated. Scientists who wish to contribute to these policy debates are naturally pulled into the role of "Issue Advocate," marshalling the science in support of a narrowed range of politically-supported options. Although this is a useful role, Pielke warns, scientists often drift into it unwittingly. As they deny any political influence on their scientific judgments, these "stealth issue advocates" can damage the authority of science even as they obscure the true nature of the political decision.
To address this problem, Pielke pleads for more scientists to act as "Honest Brokers of Policy Alternatives," to give his complete description. Such scientists, presumably as part of multi-disciplinary committees like the now-defunct Congressional Office of Technology Assessment, would act to expand the available policy alternatives rather than restrict them. Unlike the science arbiter, Pielke's honest broker recognizes an inseparability of policy issues from the corresponding scientific issues, but nonetheless provides a palette of options that are grounded in evidence.
In my technology research, I've had my own complaints about the analogous linear model. I've found that pure research often leads to more research, rather than to the promised applied research and products that make everyone's lives better. But Pielke's criticism of the linear model is more fundamental. He correctly notes that in many complex situations, scientific knowledge does not, on its own, determine a policy outcome. But he then seems to conclude that there is no legitimate role for objectively valid science that can narrow policy options.
In discussing Bjørn Lomborg's The Skeptical Environmentalist, for example, Pielke says "Followers of the linear model would likely argue that it really does matter for policy whether or not the information presented in TSE is 'junk' or 'sound' science." He then shows that for some criticisms of the book, the validity of the science was irrelevant to policy. But many of the standard talking points raised by global-warming skeptics are well within the bounds of science, so clarifying them is a useful narrowing of options, even if it doesn't lead to a single, unanimously correct policy.
Nonetheless, Pielke's short, readable book provides a helpful guide for what we can hope for in policy debates involving science, and how scientists can most productively contribute. What we can't hope for is a single, science-endorsed answer to complex issues that trade off competing interests and conflicting values. For that, we have politics.
Wednesday, November 25, 2009
Green Computing
Supercomputers run the vast simulations that help us to better predict climate change--but they also contribute to it through their energy consumption.
The lifetime cost of powering supercomputers and data centers is now surpassing the cost of buying the machines in the first place. Computers, small, medium, and large, have become a significant fraction of energy consumption in developed countries. And although the authors of Superfreakonomics may not understand it, the carbon dioxide used to supply this energy will absorb, during its time in the atmosphere, some 100,000 times more heat energy than that.
To draw attention to this issue, for the last two years researchers at Virginia Tech have been reordering the Top500 list of the fastest supercomputers, ranking them according their energy efficiency in the Top Green500 list. I have a wee news story out today on the newest list, released last Thursday, on the web site of the Communications of the Association for Computing Machinery. The top-ranked systems are from the QPACE project in Germany, and are designed for quantum chromodynamics calculations.
Calculating efficiency isn't as straightforward as it sounds. The most obvious metric is the number of operations you get for a certain amount of energy. This is essentially what Green500 measures in its MFLOPS/W, since MFLOPS is millions of floating-point operations per second and watts is joules per second.
As a rule, however, this metric favors smaller systems. It also favors slower operation, which is not what people want from their supercomputers. Some of the performance lost by running slowly can be recovered by doing many operations in parallel, but this requires more hardware. For these reasons, the most efficient systems aren't supercomputers at all. The Green500 list works because they only include the powerhouse machines from the Top500 list, which puts a floor on how slowly the competing machines can go.
Over the years, researchers have explored a family of other metrics, where the energy per operation is multiplied by some power of the delay per operation: EDn. But although these measures may approximately capture the real tradeoffs that systems designers make, none has the compelling simplicity of the MFLOPS/W metric. This measure also leverages the fact that supercomputer makers already measure the computational power to get on the Top500 list, so all they need to do extra is measure the electrical power in a prescribed way.
These systems derive much of their energy efficiency from the processor chips they use. The top systems in the current list all use a special version of IBM's cell processor, for example. I worked on power reduction in integrated circuits more than a decade ago--an eternity in an industry governed by Moore's Law--and some of my work appeared in a talk at the 1995 International Electron Devices Meeting. I also served for several years on the organizing committee of the International Symposium on Low Power Electronics and Design, but I'm sure the issues have advanced a lot since then.
In addition to the chips, the overall system design makes a big difference. The QPACE machine, for example, serves up as about half again as many MFLOPS/W as its closest competitor by using novel water-cooling techniques and fine-tuning the processor voltages, among other things. These improvements aren't driven just by ecological awareness, but by economics.
There's still lots of room for improvement in the energy efficiency of computers. I expect that the techniques developed for these Cadillac systems will end up helping much more common servers to do their job with less energy.
Tuesday, November 24, 2009
Happy Anniversary
One hundred fifty years ago today, the first edition of Charles Darwin's masterpiece On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life was published.
I picked up a copy of the book a few years ago for about $10, when the American Museum of Natural History in Manhattan had a Darwin exhibit. The most memorable display for me was the handwritten notebook entry where he first speculated about the tree-like connectivity between different species. I was humbled to be within a few feet of this tangible record of his world-changing inspiration, written with his own pen.
The book itself was very readable, intended as it was for an audience far beyond specialists. Starting with what would then have been familiar techniques of plant and animal breeding--"artificial selection"-- Darwin proposes conceptually extending that process to nature. Combining natural variation with its heritability, a Malthusian appreciation of the struggle to survive and an awareness of the immensity of geographic time, this extension seems eminently reasonable.
And yet there are challenges. Rather than bluster through them, Darwin addresses them head on, conveying an honesty and openmindededness that is bracingly refreshing in our argumentative times.
He confronted head-on, for example, the intellectual challenges of the intricate structure of the eye, fully admitting that the theory demanded that at every step of evolution there be some function for the intermediate forms. Even today, intelligent design proponents profess to be flummoxed by the very challenges that Darwin faced--and faced down.
Darwin also clearly described the tradeoffs needed for the evolution of traits like altruism, avoiding the temptation to invoke the "good of the species." To persist, such traits must provide an advantage to the group that exceeds the cost to individuals. This clear statement of the constraints of group selection needs wider appreciation today.
In these and many other areas, Darwin anticipated and addressed the confusing aspects of his explanation for evolution. And he did it all without even the benefit of Mendel's laws of genetics.
Time and again in the intervening decades, newly uncovered evidence from biology and paleontology has reinforced the essential correctness of Darwin's framework. The laws of genetics and of their DNA mechanism, the fossil record of transitional forms, and mathematical models have all confirmed and clarified the power of undirected selection of random variation for driving innovative new possibilities.
There are caveats, of course. Non-inherited mechanisms of genetic transfer change the story in important ways, especially near the single-celled trunk of the tree of life. Such revisions are hardly surprising after 150 years of scientific advancement.
What is humbling is the persistent soundness of the essence of Darwin's vision, and of this amazing book.
Sunday, November 22, 2009
Rules or Consequences
Can we learn about one phenomenon by studying a completely different one?
Putative "laboratory versions" of exotic phenomena appear regularly in the news, such as microwave analogs of "rogue" ocean waves, optical-fiber analogs of rogue waves and black holes, and, as I've discussed here, magnetic-crystal analogs of magnetic monopoles.
But not all of these experiments are equally illuminating. Researchers, and journalists who write about them, need to think clearly about how the two systems are related, and what's missing. Experiments on a model system can show what behavior arises from shared underlying rules, and how that behavior changes as conditions change. But only experiments on the original system can test whether those rules are relevant.
The results of known mathematical rules aren't always obvious. Even Newton's second law, which relates the force on an object to its acceleration, only stipulates a differential equation that researchers must solve to find how an objects position changes with time. When the force is constant, this is easy: the position follows a parabolic course in time.
For more complicated situations, scientists often can't relate the rules to the end result. In some cases they turn to simulations, which can be regarded as a model system that, ideally, embodies the mathematical rules perfectly. But simulations are often restricted to unrealistically small systems that could behave differently than the real McCoy.
In these cases, researchers can learn from actual systems that--they think--follow similar rules. For one thing, this may make precision measurements easier. Placing a block on an inclined plane, for example, slows down its acceleration due to gravity, making it possible to test the parabolic law more precisely.
Unfortunately, the model system may introduce complications of its own. The friction on a sliding block is significantly different than that air friction on a falling body--for example it's much larger before the block starts to move. Even though the rules of gravitational force are the same, the differences may completely obscure the relationship between the two systems. Researchers must then spend a lot of energy tracking down these differences.
But to draw any parallel between two systems, researchers must establish that both are governed by similar rules. Unless they know that, seeing a particular behavior in a model system, by itself, is irrelevant for deciding if the original system follows the same rules. The way to test that--but not prove it--is to do experiments on that system, and see if the behavior is similar.
In our example, if an object follows a parabolic time course, it might well be that it is responding to a constant force. (Of course, it may just be moving through curved spacetime.) With luck, the model system--the inclined plane--would have demonstrated something close to this parabolic result, even if the equations had been unsolvable. The model system then hints at a similarity of the governing rules.
Similarly, a chaotic microwave cavity or an optical fiber might show a "long tail" in the distribution of wave heights that mathematically resembles that which is experimentally measured on the ocean, and which occasionally spawns mammoth rogue waves. Because it's easier to vary the conditions in the laboratory, these experiments might also show what aspects of wave propagation are relevant to rogue-wave formation. In these systems, researchers already understand the basic features of wave propagation--the question is what happens when they combine the ingredients in various ways.
In contrast, physicists do not know whether the basic equations of physics allow magnetic monopoles. Some grand unified theories predict them, but they've never been seen in free space, despite extensive experiments. The observation of monopole excitations at low temperatures in magnetic materials called spin ices has absolutely no implications for the nature of the fundamental equations. It may be that it helps to understand how "real" monopoles would behave, if they exist. But it says nothing about whether they do.
Model systems can reveal important relationships between models and behavior. They can also uncover real-world complications that need to be included to make models more relevant. But to find out whether a model applies to a particular system in the first place, researchers need experiments on that system. Experiments on a model system aren't enough.
Thursday, November 19, 2009
Denialism
As someone who communicates science for a living, I frequently struggle to understand the widespread distrust of scientific evidence in public and private decisions. I was looking forward to some enlightenment in The New Yorker writer Michael Specter's new book, Denialism: How Irrational Thinking Hinders Scientific Progress, Harms the Planet, and Threatens Our Lives.
I was disappointed. The book scarcely addresses the origins of denialism, or even, as the subtitle advertises, its consequences. Instead, it reads as a cobbled-together series of feature articles, all too long to be called vignettes. The pieces are mostly interesting, well researched and well written, but they include a lot of background material that is peripheral to denialism. As to where the attitude comes from, Specter offers only speculation.
Specter is unlikely to make many converts to "rational thinking," since he frequently comes across as a cheerleader for progress, even as he acknowledges its risks and uncertainties. For example, near the close of his 21-page introduction, he shares a letter from a New Yorker reader: "…the question remains, will this generation of scientists be labeled the great minds of the amazing genetic-engineering era, or the most irresponsible scientists in the history of the world? With the present posture of the scientific community, my money, unfortunately, is on the latter." I regard this is a valid question, but Specter dismisses it: "Those words might as well have been torn from a denialist instruction manual: change is dangerous; authorities are not to be trusted; the present 'posture' of the scientific community has to be one of collusion and conspiracy." He doesn't seem to allow for reckless overconfidence.
Specter doesn't address climate change, which is the only big issue where denialism (as opposed to progress) threatens to "harm the planet." Cynics will note that it's also the issue where denialism promotes corporate interests, rather than opposing them. But the various chapters cover a wide range of topics.
In Vioxx and the Fear of Science, Specter reviews Merck's coverup of the heart risks of their pain medication, Vioxx. This sorry episode has been discussed elsewhere, for example in Melody Peterson's Our Daily Meds, but on its face it has little to do with irrational denial. In fact, in this case, distrust of pharmaceutical companies and the FDA are quite well founded. But in the final section of the chapter that reads like an afterthought, Specter blames much of the public's disregard for scientific evidence such betrayals of trust, although he gives little evidence for this connection.
Specter also uses the Vioxx case to illustrate a common problem: undue attention to acute harms rather than small, distributed benefits. He even argues that the thousands of deaths from Vioxx might have been a reasonable price to pay for its pain relief benefits to millions. Such weaknesses in risk assessment certainly skew many policy and private decisions. But our oft-lamented poor balancing of accepted risks and benefits strikes me as somewhat distinct from denialism, in which scientific evidence for benefit or harm is dismissed entirely.
Both denialism and poor weighing of pros and cons also come to play into the second chapter, Vaccines and the Great Denial. Specter makes it clear there is virtually no science supporting the anti-vaccine movement, and documents the highly misleading selective quotation of a government report in Robert Kennedy's famous Rolling Stone story. This is an easy case to make, but he does it convincingly.
In The Organic Fetish, Specter combines two distinct food-related issues. He shows convincingly that the benefits of "organic" foods are less clear-cut than advocates would like to believe, although I prefer Michael Pollan's wonderful book, The omnivore's dilemma: a natural history of four meals. But Specter's denialism them lets him zig-zag erratically between organic and genetically modified (GM) foods. He compelling despairs over African nations' rejecting GM foods for their starving populations, but he is too willing to accept the standard, long disproven reassurances about the limited spread of modified foods. Still, Specter resoundingly dispels the mythical distinction between modern modifications and those that have been accepted for decades or millennia.
Specter's chapter on food supplements and alternative medicine, The Era of Echinacea, also has an easy target, although he notably includes multivitamin supplements among the snake oils. But again, his discussion lacks a clear explanation of why many people trust these uncontrolled additives more than they do the tightly-regulated products of the pharmaceutical industry.
Race and the Language of Life combines two disparate topics. Specter's discussion of the complex role of genetics in disease is impressively thorough and accurate, and he gives it a human touch with his own genetic testing. But he also invokes the importance of genetics to support the use of race in medicine. Although Specter is no doubt correct that race is often avoided for political reasons, there is a legitimate scientific question that he fails to clarify: how much of the genetic variation in medical response can be explained with traditional notions of race? If within-group variation is large and the differences between groups are largely statistical, the divisive introduction of race may bring little benefit. The messy story behind the heart drug BiDil, approved by the FDA for African Americans, for example, makes it unconvincing as his poster child for race-based medicine.
Surfing the Exponential delves into the nascent field of synthetic biology, covering much the same ground as Specter's recent story in The New Yorker. This chapter is rich in technical detail on the promise of the technology, and to a lesser degree with the risks of making new, self-replicating life forms. Ultimately, though, Specter advocates "a new and genuinely natural environmental movement--one that doesn't fear what science can accomplish, but only what we might do to prevent it."
Such denialism is no more defensible for assessing risks than for judging benefits--both should to be analyzed thoroughly. Fear of the unknown is not always irrational.
Tuesday, November 17, 2009
New Guidelines for Breast-Cancer Screening
A few weeks ago, as I reported here, Gina Kolata at the New York Times reported that the American Cancer Society was planning to scale back their recommendations on routine screening for prostate and breast cancers.
As discussed by the Knight Tracker, she got a lot of grief for this story, and the next day the Times published a more reserved follow-up story by Tara Parker-Pope, also discussed by the Tracker. In fact, her primary source at the society, Dr. Otis Brawley, later wrote a letter to the editor denying any intention to change the guidelines (although he is on record cautioning about overscreening).
This isn't the first time that a page-one story by Kolata has gotten into trouble. Her 1998 story on cancer drugs was cited as a cautionary tale in my medical-writing course at NYU. That story quoted James Watson as saying (privately, at a banquet) that Judah Folkman was "going to cure cancer in two years" with his amniogenesis inhibitors. Watson later denied saying any such thing.
Nonetheless, Kolata accurately conveyed a painful dilemma of cancer screening: more isn't necessarily better. Not for all cancers, and not for all patients.
The U.S Preventive Services Task Force has now issued revised recommendations for breast-cancer screening for patients who have no indications of high risk. In part, they moved the earliest age for mammography back up from 40 to 50, at which point they recommend a scan every two years rather than every year.
These recommendations were based not primarily on financial costs, but on health risks to patients:
"The harms resulting from screening for breast cancer include psychological harms, unnecessary imaging tests and biopsies in women without cancer, and inconvenience due to false-positive screening results. Furthermore, one must also consider the harms associated with treatment of cancer that would not become clinically apparent during a woman's lifetime (overdiagnosis), as well as the harms of unnecessary earlier treatment of breast cancer that would have become clinically apparent but would not have shortened a woman's life. Radiation exposure (from radiologic tests), although a minor concern, is also a consideration."
The blog, Science-Based Medicine, has a thoughtful and thorough discussion of the issue, written before the recent recommendations. I highly recommend it.
In a rather odd move, the Times published a balancing article by Roni Caryn Rabin on the same day (at least in the paper edition), although it was buried in the "Health" section, not on the front page with Kolata's.
Rabin's story empathetically interviews screening advocates, including people who have been treated for breast cancer. But in its empathy, story misses the opportunity to clarify the issues. Or perhaps in the extended quotes, the author is deliberately allowing the sources to reveal themselves? It's hard to tell.
For example, one woman calls screening her "security blanket." "'If someone ran a computer analysis that determined that wearing a seat belt is not going to protect you from being killed during a crash, would you stop using a seat belt?' Ms. Young-Levi asked."
Although it's hard to imagine, I certainly would stop using a seat belt if the best evidence indicated it, whatever psychological security I might ascribe to it.
The story later quotes another survivor: "'You're going to start losing a lot of women,' said Sylvia Moritz, 54, of Manhattan, who learned she had breast cancer at 48 after an annual mammogram. 'I have two friends in their 40s who were just diagnosed with breast cancer. One of them just turned 41. If they had waited until she was 50 to do a routine mammogram, they wouldn't have to bother on her part — she'd be dead.'"
The author negligently lets this quote stand: the whole point of the recommendations is that if those friends had not been diagnosed, they might be doing just fine now, without the risk of the tests and procedures they underwent because of the diagnosis.
But the unfortunate reality is that we need tests that better predict cancer progression, rather than merely signaling its presence. Without such tests, the recommendations can only trade off lives lost (and other damage) because treatment was unnecessarily aggressive with other lives lost because it wasn't aggressive enough.
Monday, November 16, 2009
That's a Wrap
Almost every human cell contains DNA that, if stretched out, would extend several meters. Packing that length into a nucleus perhaps 1/100,000 of a meter in diameter--and unpacking it so it can be duplicated when the cell divides--is an almost miraculous feat.
In addition, although the details of the packing are not fully understood, it is clear that different parts of the DNA are packed differently, and this packing strongly influences how readily its sequence is transcribed. The packing provides yet another mechanism for regulating gene expression.
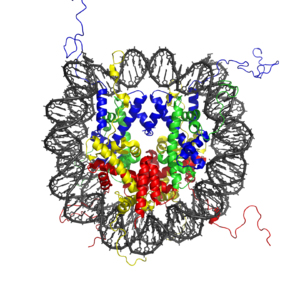
To form a nucleosome, DNA wraps almost twice around a group of eight histone proteins, two copies each of H2A (yellow), H2B (red), H3 (blue), and H4 (green). Chemical modifications of the stray tails of the histones affect the packing of nucleosomes with one another and modify transcription of the neighboring DNA. (Higher resolution at the Wikipedia Description Page.)
At the smallest level of organization, the DNA double helix winds around an octet of histone proteins to form what's collectively called a nucleosome. Usually the nucleosomes are quite densely arrayed along the DNA, connected by short linking segments of DNA like "beads on a string."
The nucleosomes tend to jam together as closely as possible. But they form at some DNA sequences more easily than others, due to details of the attraction of different bases to the histones and the need to bend the double helix. They also compete for binding with DNA-binding proteins like transcription factors and the proteins that initiate transcription.
Using the known sequence to model the competition among nucleosomes and between nucleosomes and proteins is a complex statistical-mechanics challenge that has attracted the attention of biophysicists, as discussed in a symposium I covered for the New York Academy of Sciences a couple of years ago.
The exact position matters. The enzyme that transcribes genes, RNA polymerase II, needs access to the DNA, whose the two strands must be temporarily separated to get access to the sequence to copy it. The wrapping of DNA around histones slows both the initiation of transcription and the continued elongation of the RNA transcript.
Still, wrapping around histones is only the first level of DNA packing. The way that the resulting nucleosomes assemble into larger structures is even more important in determining which DNA sections are actively transcribed.
Friday, November 13, 2009
Lies, Damn Lies, and…
Statistics don't lie. People do.
I have the greatest respect for statisticians, who methodically sift through messy data to determine what can confidently and honestly be said about them. But even the most sophisticated analysis depends on how the data were obtained. The miniscule false-positive rate for DNA tests, for example, is not going to protect you if the police swap the tissue samples.
One of the core principles in clinical trials is that researchers specify what they're looking for before they see the data. Another is that they don't get to keep trying until they get it right.
But that's just the sort of behavior that some drug companies have engaged in.
In the Pipeline informs us this week of a disturbing article in the New England Journal of Medicine. The authors analyzed twenty different trials conducted by Pfizer and Parke-Davis evaluating possible off-label (non-FDA-approved) uses for their epilepsy drug Neurontin (gabapentin).
If that name sounds familiar, it may be because Pfizer paid a $0.43 billion dollar fine in 2004 for illegally promoting just these off-label uses. As Melody Peterson reported for The New York Times and in her chilling book, "Our Daily Meds," company reps methodically "informed" doctors of unapproved uses, for example by giving them journal articles on company-funded studies. The law then allows the doctors to prescribe the drug for whatever they wish.
But the distortion doesn't stop with the marketing division.
The NEJM article draws on internal company documents that were discovered for the trial. Of 20 clinical trials, only 12 were published. Of these, eight reported a statistically significant outcome that was not the one that was described in the original experimental design. The authors say "…trials with findings that were not statistically significant (P≥0.05) for the protocol-defined primary outcome, according to the internal documents, either were not published in full or were published with a changed primary outcome."
A critical reason to specify the goals, or primary outcome, ahead of time is that the likelihood of getting a statistically significant result by chance increases as more possible outcomes are considered. In genome studies, for example, the criterion for significance is typically reduced by a factor that is the number of genes tested, or equivalently the number of possible outcomes.
None of this would be surprising to Peterson. She described a related practice in which drug companies keep doing trials until they get two positive outcomes, which is what the FDA requires for approval.
By arbitrary tradition, the numerical threshold for statistical significance is taken as a 5% or less chance that an outcome arose by chance (P-value). This means that if you do 20 trials you'll have a very good chance of getting one or more that are "significant," even if there is no effect.
A related issue arose for the recent, highly publicized results of an HIV/AIDS vaccine test in Thailand. Among three different analysis methods, one came up with a P-value of 4%, making it barely significant.
This means is that only one in twenty-five trials like this would get such a result by chance. That makes the trial a success, by the usual measures.
But this trial is just one of many trials for potential vaccines, most of which have shown no effect. The chances that any one of these trials gave a positive result is much larger, presumably more than 5%.
In addition, the Thai vaccine was expected to work by slowing down existing infection. Instead, the data show reduced rates of initial infection. Measured in terms of final outcome (death), it was a success. But in some sense the researchers moved the goalposts.
Sometimes, of course, a large trial can uncover a real but anticipated effect. It makes sense to follow up on these cases, recognizing that a single result is only a hint.
Because of the subtleties in defining the outcome of a complex study, there seems to be no substitute for repeating a trial, stating a clearly defined outcome. Good science writers understand this. It would be nice to think that the FDA did, too, and established procedures to ensure reliable conclusions.
Thursday, November 12, 2009
Guilt by Association
Many of the molecular transformations in cells occur inside of complexes, each containing many protein molecules and often other molecules like RNA.
Determining which molecules are in each complex is a critical experimental challenge for unraveling their function.
Ideally, biologists would identify not just the components, but the way they intertwine at an atomic level, for example using x-ray crystallography. The Nobel-prize-winning analysis of the ribosome showed that this detailed structural information also illuminates how the pieces of the molecular machine interact to carry out its biochemical task.
But growing and analyzing crystals takes years of effort. In many cases researchers are happy just to know which molecules are in which complexes. As a first step, biologists have developed several clever techniques to survey thousands of proteins to see which pairs interact, and to confirm whether those interactions really happen in cells.
Identifying additional protein members of complexes requires chemical analysis like chromatography and increasingly powerful mass spectrometry techniques. In contrast, to explore how DNA and RNA act in complexes, researchers can take advantage of the sequence information available for humans and most lab organisms.
To find out which DNA regions bind with a particular protein transcription factor, for example, biologists use Chromatin ImmunoPrecipitation, or ChIP. Bound proteins from a batch of cells are chemically locked to the DNA with a cross-linker like formaldehyde.
This technique then requires an antibody that binds only to the protein (and its bound DNA), and which is sooner or later tethered to a particle. After breaking apart the DNA, the particle precipitates to the bottom of the solution carrying ("pulling down") its bound molecules, which are then separated and analyzed.
A related technique identifies proteins bound to an antibody-targeted partner. Ideally, the methods identify components that were already bound just before the cells are broken up to begin the experiment, rather than all possible binding sites, so they flag only biologically relevant pairings.
For DNA, the state of the art until recently was "ChIP-chip," which uses microarrays to try to match the pulled down DNA to one of perhaps a million complementary test fragments on an analysis "chip." The advent of high-throughput sequencing has allowed "ChIP-seq," in which the sequence of the bound DNA is directly measured and compared by software to the known genome to look for a match. This was the method used recently to find enhancer sequences by their association with a known enhancer-complex protein.
A similar method can identify the RNA targets of RNA-binding proteins, as discussed by Scott Tenenbaum of the University at Albany at a 2007 meeting that I covered for the New York Academy of Sciences (available through the "Going for the Code" link on my website's NYAS page).
Once the fragments are identified, researchers can try to dissect the elements of the sequence that make them prone to binding by a particular protein. When successful, this procedure allows them to identify other targets for interaction with proteins using only computer analysis of sequence information. These bioinformatics techniques are a critical time saver, because the experiments show that each protein can bind to many different molecules in the cell.
Experiments like these are revealing many of the intricate details of cellular regulation, but also how much more there is to learn.
Wednesday, November 11, 2009
Enhancers, Insulators, and Chromatin
Some DNA regions affect the activity of genes that are amazingly far away in the linear sequence of the molecule.
The best known way that genes turn on and off--and thus determine a cell's fate--is when special proteins bind to target DNA sequences right next to different genes--within a few tens of bases. Together with other DNA-binding proteins, these sequence-specific transcription factors promote or discourage transcription of the sequence into RNA. This mechanism is an example of what's called cisregulatory action, because the gene and the target sequence are on the same molecule.
There's another type of sequence that affects genes on the same DNA molecule, but these can be so far away--tens of thousands of bases--that it seems odd to call them cis-regulatory elements. These "enhancers" can be upstream or downstream of the gene they regulate, or even inside of it, in an intron that doesn't code for amino acids. In fact, some of them affect genes on entirely different chromosomes.
Enhancers have important roles in regulating the activity of genes during development, "waking up" in specific tissues at specific times.
The flexible location makes it hard to find enhancers in the genome, and researchers have also struggled to find clear sequence signatures for them. In ongoing work that I described last year for the New York Academy of Sciences, Eddy Rubin and his team at Lawrence Berkeley Labs instead looked for sequences that were extremely conserved during evolution. Although evolutionary conservation is not a perfect indicator, when they attached a dye near these sequences in mice, they often found telltale coloration appearing in particular tissues as the mouse embryos developed.
Years of effort have uncovered many important clues about how enhancers exert their long-distance effects, but still no complete picture. Most researchers envision that the DNA folds into a loop, bringing the enhancer physically close to the promoter region at the start of a gene. Proteins bound to the enhancer region of the DNA, including sequence-specific proteins that can also be called transcription factors, can then directly interact with the proteins bound near the gene, and enhance transcription of the DNA.
But the enhancement effect can be turned off, for example, when researchers insert certain sequences in the DNA sequence between the gene and the enhancer. These "insulator" sequences seem to restrict the influence of the enhancer to specific territories of the genome. Naturally occurring insulators serve the same restrictive function, but they can also be turned off, for example by chemical modification, providing yet another way to regulate gene activity.
If enhancers work by looping, it seems surprising that an intermediate sequence could have such a profound effect. Researchers have proposed various other explanations as well.
In addition to stopping the influence of enhancers, many insulators restrict the influence of chromatin organization. Biologists have long recognized that the histone "spools" that carry DNA can pack in different ways that affect their genetic activity. In a simplistic view, tight packing makes it hard for the transcription machinery to get at the DNA. This chromatin packing can be modified in the cell, and is one important mechanism of epigenetic effects that persistently affect gene expression even through cell division.
As a further confirmation of the close relationship, Bing Ren of the Ludwig Institute and the University of California at San Diego has successfully used known chromatin-modifying proteins to guide him to enhancers, in work that I summarized from the same meeting last year.
One model that combines some of these ingredients says that loops of active DNA are tethered to some fixed component of the nucleus, and that enhancers can only affect genes on the same loop. If insulators act as tethers, this naturally explains how it limits interactions to particular regions (which are then lops). There is still much to be learned, but enhancers and the chromatin packing appear to be tightly coupled.
I suspect that enhancers have been somewhat neglected both because their action mechanism is so confusing and because definitive experiments have been difficult. But recent experiments done in a collaboration between Rubin's and Ren's teams have used a protein called p300, which binds to the enhancer complex, to identify new enhancers with very high accuracy. Moreover, the binding changes with tissue and development just as the enhancer activity does. These and other experiments are opening new windows into these important regulatory elements.
Tuesday, November 10, 2009
Free Will and Quantum Mechanics
[NOTE: This piece is modified from one written in the spring of 2005, but never published because it was too demanding, so be warned.]
In 2004, two mathematics professors from Princeton University devised the simplest proof yet that the world really is unpredictable at a microscopic level.
Quantum mechanics has passed many experimental tests, but it generally predicts only the probabilities of various outcomes. Over the decades, many physicists, notably Einstein, have longed for a description that doesn't involve "throwing dice."
The Princeton "Free-Will Theorem" concludes that, if experimenters can make choices freely, then this unpredictable behavior of elementary particles is unavoidable. But other experts suspect that the result is another manifestation of the "spooky action at a distance," that dominates the quantum world.
"Physicists usually are not impressed much," admitted John Horton Conway, the inventor of the 1970 cellular-automaton game he called Life. "They actually believe quantum mechanics." Indeed, few dispute that quantum mechanics gives correct predictions. But Conway and his colleague Simon Kochen said that although their conclusions are familiar, they start with three axioms, called SPIN, TWIN, and FIN, that are much simpler than previous theorems, and avoid "counterfactual" experiments that can't be done.
The first axiom, called SPIN, is based on an unusual property of a "spin-one" elementary particle: Measuring the square of its angular momentum, or spin, as projected along three perpendicular directions will always yield two ones and one zero. This bizarre property is usually derived from quantum mechanics, but it could have been observed independently.
"We don't have to know what 'the square of the spin' means," Conway said. "It's really rather important that we don't, because the concept 'squared spin' that we're asking about doesn't exist-- that's one of the things that's proved."
If such a squared spin existed, then experimenters could, in principle, choose a direction to measure it along and know in advance whether it would be one or zero. But in a groundbreaking theorem published in 1967, Kochen and E.P. Specker showed that it is impossible to prepare a list beforehand that gives the required two ones and a zero for all possible sets of measurement directions. They concluded that there are no "hidden variables" that describe the "real" spin.
Later researchers, however, realized that such hidden variables could logically exist, but only if their values changed depending on which measurements were chosen, a property known as "contextuality."
To avoid this problem, Conway and Kochen analyzed pairs of particles with matched properties. Their second axiom, which they call TWIN, is that experimenters can make and separate such pairs. This ability is well established, and experiments on the pairs have confirmed the quantum prediction that measurements of their properties remain correlated long after they separate. (I described one recent experiment for Technology Review.)
In 1964, John Bell showed that the some measurements on the two particles can only be explained if each particle somehow continues to be affected by the other, even though they are far apart. This surprising "nonlocality" has since been confirmed in numerous experiments, which find that the correlation, averaged over many pairs, exceeds the maximum for any conceivable local theory.
To avoid the need for statistical averages, Conway and Kochen applied the Kochen-Specker theorem to a single, matched, spin-one pair. If researchers measure the same component of the squared spin for both particles, they should always find either both zeroes or both ones. (Rutgers student Douglas Hemmick also derived this result in his 1996 doctoral thesis.) A similar "Bell's theorem without inequalities" was described in 1989 by Daniel Greenberger, Michael Horne, and Anton Zeilinger, using three "spin-1/2" particles.
Conway and Kochen's third axiom, FIN, is grounded in special relativity, and says that information travels no faster than, say, the speed of light. In spite of appearances, they say, nonlocal effects do not exceed this speed limit, because they describe only coincidence between two measurements, not causation. In fact, special relativity makes it meaningless to say that either of two widely separated measurements occurred "first," so it makes no sense to talk of information passing between the two.
Combining these ingredients, Conway and Kochen imagine that the squared spin is measured in all three directions for one member of a pair. If an experimenter measures the spin of the other member along any of these directions, the result must agree with the one for the first member.
But if the experimenter is free to choose which direction to measure, then because of FIN, that information is not available to the first particle. The result of the first measurement can't depend on her choice, but since there is no way to consistently anticipate all possible measurements, Conway and Kochen conclude that no hidden variable could have predicted the outcome. The only way to avoid this unpredictability, they say, is if the experimenter wasn't really free to choose which experiment to do.
Tim Maudlin, who heard Conway present the work in a colloquium in November 2004, disputes that conclusion. A philosophy professor at Rutgers University and author of "Quantum Non-Locality & Relativity," Maudlin remarked that saying the behavior of a particle cannot be determined by information in its own past "is just what we mean by non-locality," which is already clearly established. "You've taken the contextuality and stretched it out" to include both members of the pair, he asserts.
Conway and Kochen published their Free Will Theorem in 2008, and the Princeton Alumni Weekly has posted videos of lectures by Conway. But it appears that other scientists are free to choose whether to believe it.
Monday, November 9, 2009
The Wisdom of Ignorance
Physics and math demand a certain mode of thought. Lots of people think that doing well in those classes takes intelligence, and that's part of it. But they also require something else that is not always a good thing: comfort with abstraction, or stripping problems down to an idealized cartoon.
Those of us who excelled in these subjects can be a bit smug towards those who didn't, but replacing real life with a cartoon isn't always a good thing. In addition to hindering social relations, it can obscure important truths.
It's interesting to contrast Aristotle's view, for example--that objects in motion naturally come to rest--with Newton's--that they naturally keep moving. Thinking about familiar objects, you have to grant that Aristotle had a good point. Of course, he'll leave you flat: if you figure out how to include friction, Newton is going to get you a lot further--even to the moon. But beginning students are asked to commit to an abstract formalism that has a stylized and flawed relationship to the world they know.
Probability has a similar problem.
Most normal people, for example, expect that after a flipped coin shows a string of heads, tails is "due." Probability theory says otherwise: the coin doesn't "know" what happened before, so the chances on the next flip are still 50/50. Abstraction wins, intuition loses.
[Actually, Stanford researchers showed in 2007 (pdf here) showing that unless a coin is flipped perfectly, the results will not be 50/50: if the coin is spinning in its plane at all, its angular momentum will tend to keep it pointed the way it started out. But that's a minor issue.]
On the other hand, there are lots of cases where common intuition is "directionally correct." Take another staple of introductory probability courses: a bag full of different-colored balls. In this case, the probability won't stay the same unless you put each ball back in the bag after you choose it. If you keep it, choosing a ball of one color will increase the chances of a different color on the next pick, in keeping with intuition.
Of course, intuition doesn't get the answer with any precision, and it gets it completely wrong for the coin flip. To do it right, you need the abstract formalism. Still, it's easy to imagine that our brains are hard-wired with an estimating procedure that gets many real-world cases about right.
In other cases, our intuition is more flexible than slavish devotion to calculation. Suppose I start flipping a coin. It's not surprising to see heads the first time, and the second time. How about the third time? The tenth? If it keeps coming up heads, you will quickly suspect that there's a problem with you original assumption that the probability is 50%. Your natural thought processes will make this shift naturally, even if you might be hard pressed to calculate why. Probability theory is not going to help much when the assumptions are wrong.
It's true that people are notoriously bad at probability. ScienceBlogger Jason Rosenhouse has just devoted an entire book to one example, the "The Monty Hall Problem: The Remarkable Story of Math's Most Contentious Brain Teaser. (It was also discussed in 2008's The Drunkard's Walk, by Leonard Mlodinow, and in The Power of Logical Thinking, by Marilyn Vos Savant (1997), the Parade columnist who popularized it.)
The Daily Show's John Olliver amusingly explored how simple probability estimates (especially at around minute 3:20) help us misunderstand the chances that the Large Hadron Collider will destroy the world.
Still, our innate estimation skills developed to deal tolerably well with a wide variety of situations in which we had only a vague notion of the underlying principles. Highly contrived, predictable situations like the coin flip would have been the exception. Even though our intuition frequently fails in detail, it helped us survive a complex, murky world.
Friday, November 6, 2009
Group Selection
Can evolution select attributes "for the good of the group," even if they're bad for individuals? That is the essential notion of group selection, which has been a highly controversial area of evolutionary theory. In fact, some evolutionary biologists insist it never happens.
David Sloan Wilson of Binghamton University disagrees, in an interesting series that he calls Truth and Reconciliation in Group Selection. Originally at Huffington Post, he's re-posting the series in his new blog location at ScienceBlogs.
The author of the very readable Evolution for Everyone: How Darwin's Theory Can Change the Way We Think About Our Lives, Wilson is a long-standing proponent of group selection in some circumstances. But his chosen title evokes his contention that opposition to even the possibility of group selection has become a kind of dogma, in stark contrast to the ideal of scientific hypothesis testing.
From the time Darwin introduced it until the 1960s, Wilson says, group selection was taken seriously. But then W.D. Hamilton devised a model that explained why evolution would favor acts that benefit close relatives. As long as the benefit to the relatives, multiplied by the fraction of shared genes, exceeds the cost to the individual, he argued, an action would make propagation of the genes more likely.
But a successful model based on kinship doesn't rule out the possibility of other successful explanations.
Clearly, group selection requires that the group advantage mathematically outweigh the cost to the individual. So the details matter; the existence of one model in which individual needs triumph, such as John Maynard Smith's influential "haystack model," doesn't prove that group needs would not triumph under other circumstances. Nonetheless, Wilson says, the field adopted the kinship model as the only way that evolution could produce altruistic behavior.
Wilson and others have since created models in which group selection works. A theory that encompasses both kinship and other drivers of group selection was developed by the science writer George Price, as described in this 2008 article from Current Biology. Hamilton himself accepted this more general framework for balancing group and individual needs.
Experiments have also demonstrated the effect. Wilson recalls an experiment where researchers artificially selected chickens for greater egg production, according to two different criteria. They found that choosing the individual hens in a cage that laid the most eggs resulted, over generations, in less egg production than selecting cages that together laid the most eggs.
There is no question that selection "for the good of the group" has been carelessly used to motivate "just-so stories" for all sorts of traits. But it seems equally clear that there are examples of altruistic group behavior that does not derive from shared genes. Even the special teamwork that characterizes social insects, for example, still occurs in species like termites that don't have the highly-shared genes that we know from bees.
A comprehensive understanding of how evolution works in complex situations is critical to many areas of biology, such as the inference of biological function from evolutionary constraint. It is troubling to read Wilson's description of a field so mired in prejudice and tradition that it fails to fairly evaluate and incorporate valid new evidence.
Thursday, November 5, 2009
Evolution of a Hairball
Do DNA sequences evolve more slowly if they play important biological roles?
For many genomics researchers, the answer is so self-evidently "yes" that the question is hardly worth asking. Indeed, they often regard sequence conservation between species, or the evolutionary constraint that it implies, as a clear indication of biological function.
And sometimes it is a good indication. But in general, as I described in my story this summer in Science, this connection is only weakly supported by experiments, such as the exhaustive exploration of 1% of the human genome in the pilot phase of the ENCODE project. That work found that roughly 40% of constrained sequences had no obvious biochemical function, while only half of biochemically active sequences seemed to be constrained.
One reason for this is that many important functions, such as markers for alternative splicing, 3D folding of transcribed RNA, or DNA structure that affects binding by proteins, may have an ambiguous signature in the DNA base sequence.
But another reason (there's always more than one!) is that evolutionary pressure depends on biological context.
Several of my sources for the Science story emphasized that redundancy can obscure the importance of a particular region of DNA. For example, deleting one region may not kill an animal, if another region does the same thing. By the same token, redundant sequences may be less visible to evolution, and therefore freer to change over time. Biologists know many cases of important new functions that have evolved from a duplicate copy of a gene.
But redundancy, in which two sequences play interchangeable roles, is only one of many ways that genetic regions affect each other, and a very simple one at that. As systems biologists have been revealing, the full set of interactions between different molecular species forms a rich, complex network, affectionately known as "the hairball."
For some biologists, the importance of context on evolution is obvious. When I spoke on this subject on Tuesday at The Stowers Institute, for example, Rong Li pointed to the work of Harvard systems biologist Mark Kirschner. Kirschner, notably in the 2005 book The Plausibility of Life that he coauthored with John Gerhart, describes biological systems as comprising rigid core components controlled by flexible regulatory linkages.
The conserved core processes generally consist of many complex, precisely interacting pieces. They may be physical structures, like ribosome components, or systems of interactions like signaling pathways. Their structure and relationships are so finely tuned that any change is likely to disrupt their function, so their evolution will be highly constrained.
In contrast, the flexible regulatory processes that link these core components can fine-tune the timing, location, or degree of activity of the conserved core processes. Such changes are at the heart of much biological innovation. For example, the core components that lead to the segmented body plan of insects are broadly similar, at a genetic level, to those that govern our own development. Our obvious differences arise from the way these components are arranged in time and space during development.
The weak predictive power of conservation is particularly relevant as researchers comb the non-protein-coding 98.5% of the genome for new functions. Many of these non-coding DNA sequences are regulatory, so they may evolve faster. Indeed, Mike Snyder of Yale University observed a rapid loss of similarity in regulatory RNA between even closely-related species in deep sequencing studies he described at a symposium at the New York Academy of Sciences (nonmembers can get to my write-up by following the "Go Deep" link at the NYAS section of my website).
Quantifying how evolutionary pressures depend on the way genes interact is likely to keep theorists busy for years to come. But it is clear that the significance of evolutionary constraint in a DNA sequence--or its absence--depends very much on where it fits in the larger biological picture.
Subscribe to:
Posts (Atom)


